Mucopolissacaridose Tipo I
Também chamada Doença de Hurler e Doença de Scheie
As Mucopolissacaridoses (MPS) são doenças metabólicas hereditárias causadas por erros inatos do metabolismo que determinam a diminuição da atividade de determinadas enzimas, que atuam numa estrutura da célula chamada lisossomo. As MPS fazem parte de um grupo chamado Doenças de Depósito Lisossômico.
O lisossomo
Nosso organismo é feito de células, milhões delas. As células se multiplicam, vivem e morrem diariamente, sendo assim constantemente renovadas. Quando uma célula morre, é imediatamente destruída e seus componentes são reutilizados pelo organismo.
Quando uma célula é destruída, seus componentes são fagocitados ("comidos") por outras células, chamadas macrófagos, que são parentes dos glóbulos brancos do sangue. Dentro dos macrófagos, o material da célula morta fica dentro de uma espécie de "bolha", chamada lisossomo. No lisossomo, a célula introduz enzimas, que são substâncias que "digerem" os restos celulares, quebrando substâncias grandes em partes menores, que podem ser reutilizadas.
As enzimas que agem nos lisossomos são fabricadas pela própria célula, seguindo uma "receita" que fica no núcleo: os genes. Se uma pessoa sofre alguma alteração em um gene, dizemos que ocorreu uma mutação. Quando ocorre uma mutação em um gene, a "receita" fica alterada e a enzima que é fabricada com base nesta receita alterada, geralmente não funciona.
As mucopolissacaridoses
Nas MPS ocorre deficiência ou falta de enzimas que digerem substâncias chamadas Glicosaminoglicanos (GAG), antigamente conhecidas como mucopolissacarides e que deram nome à doença.
Os GAG são moléculas formadas por açúcares, que se ligam a uma proteína central, absorvem grande quantidade de água, adquirem uma consistência mucóide, viscosa, o que garante a essa estrutura uma função lubrificante e de união entre os tecidos, permitindo por exemplo o movimento das articulações (juntas) do corpo. Quando os GAG não são digeridos corretamente, devido à deficiência de alguma enzima, eles ficam depositados no interior dos lisossomos e também são eliminados pela urina.
As manifestações clínicas das MPS variam de acordo com a enzima que está em falta no portador da doença e os tipos conhecidos podem ser vistos no Quadro 1.

Mecanismo de herança
A constituição genética de cada pessoa é determinada pelos genes que recebe do pai e da mãe, sendo exatamente metade de cada um. O sexo é determinado pela presença de dois cromossomos X nas mulheres (XX) e um X e um Y nos homens (XY).
Todas as características da aparência e do metabolismo da pessoa são determinadas pela constituição genética. Cada enzima do organismo é produzida a partir da informação contida em um ou mais pares de genes, que vieram do pai e da mãe.
Dos 30.000 pares de genes que cada pessoa possui, há alguns poucos com defeitos, mas que não determinam isoladamente nenhuma doença, pois como todos os genes são em pares, um normal compensa o outro com defeito. O problema acontece quando um homem e uma mulher carregam, por acaso, o mesmo gene com defeito e podem passá-los aos filhos levando ao aparecimento de doenças genéticas.
Nas MPS dos tipos I, III, IV, VI e VII, o pai e a mãe carregam obrigatoriamente um gene com defeito cada um. Os pais são normais porque só têm um gene com defeito, sendo o outro gene do par normal.
Vamos chamar o gene normal de “A” e o com defeito de “a”, o pai é Aa e a mãe também Aa, para aquela determinada enzima que não funciona direito na doença de seu filho ou filha. Toda vez que este casal for conceber um filho existem quatro possibilidades ao acaso, da constituição da criança, tendo uma chance entre as quatro, ou seja 25%, de ter um filho ou filha com a doença (aa), como pode ser observado na Figura 1. Esta forma de herança é chamada autossômica recessiva.
Na MPS do tipo II o gene com defeito é carregado em um dos cromossomos X da mãe que, como tem dois cromossomos X, o com defeito é anulado pelo outro normal. A mãe portadora do gene da doença tem uma chance de 50% de ter um filho com a doença e 50% de ter uma filha que também pode passar o gene aos seus filhos e filhas, como pode ser observado na Figura 2. Esta forma de herança é chamada ligada ao X.

Mucopolissacaridose Tipo I
A Mucopolissacaridose I (MPS I) é uma rara doença autossômica recessiva, com manifestações patológicas em múltiplos sistemas de órgãos e tecidos. A doença é causada por um defeito na codificação genética da enzima lisossômica α-L-iduronidase; em conseqüência disso, as células das pessoas afetadas não conseguem produzir a enzima ou a produzem em pequenas quantidades. Isto resulta em uma incapacidade do lisossomo efetuar a decomposição gradual de certos glicosaminoglicanos (GAG) - a saber, sulfato de dermatan e sulfato de heparan - um processo essencial para o crescimento normal e a homeostasia dos tecidos.1 Esses glicosaminoglicanos, que são constituintes importantes da matriz extracelular, líquido das juntas e tecido conetivo em todo o corpo, acumulam se progressivamente no lisossomo, finalmente causando a disfunção da célula, do tecido e do órgão por mecanismos fisiopatológicos em grande parte desconhecidos.
A MPS I é considerada uma disfunção no armazenamento prototípico lisossômico com doença multi-sistêmica progressiva e apresentando características que variam dependendo da posição do paciente no desenvolvimento da doença.

As classificações históricas de MPS I, síndromes de Hurler, Hurler-Scheie e Scheie não refletem adequadamente a enorme variação nos sintomas clínicos. As manifestações vistas com cada classificação não são mutuamente distintas e muitas vezes se superpõem. Portanto, a doença é melhor caracterizada como deficiência de α-L-iduronidase com ou sem o envolvimento de CNS.
Sintomas Iniciais de MPS I
0-6 meses:
Rinite crônica; |
Otite média recorrente ou "infecção no ouvido médio"; |
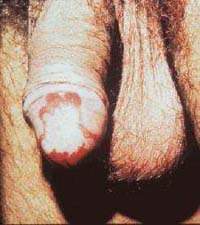
Hérnia umbilical ou inguinal; |
Crescimento e tamanho de cabeça acima do normal. |
6 meses - 12 anos:
Dismorfismos faciais; |
Hepatoesplenomegalia; |
Deformidades esqueléticas; |
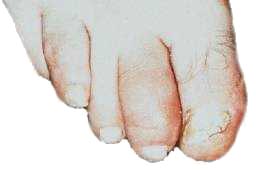
Rigidez nas juntas; |
Retardo no desenvolvimento; |
Opacidade da córnea; |
Rinite crônica; |
Otite média recorrente ou "infecção no ouvido médio". |
12+ anos:
Opacidade da córnea; |
Rigidez nas juntas; |
Doença cardíaca valvular. |
Diagnóstico
O diagnóstico de MPS I depende de ser demonstrada uma deficiência da enzima lisossômica α-L-iduronidase. A atividade desta enzima pode ser medida na maior parte dos tecidos; contudo, o diagnóstico geralmente é feito com o uso de:
leucócitos sanguíneos periféricos, |
cultura de fibroblastos, |
soro, |
plasma. |
Os achados em MPS I podem se superpor àqueles de outras disfunções do armazenamento lisossômico, principalmente outras mucopolissacaridoses e deficiência de sulfatases múltiplas. Achados clínicos e testes bioquímicos detalhados são necessários para sua diferenciação. Há alguns sinais e sintomas clínicos iniciais que por si só não são diagnósticos, mas podem justificar testes mais definitivos. Embora os achados na apresentação variem com a gravidade da deficiência, é preciso que se suspeite de MPS I em pessoas com características faciais grosseiras e hepatoesplenomegalia, assim como achados característicos do esqueleto, das juntas ou oculares.
Seu médico de confiança poderá indicar-lhe um médico geneticista, que é o especialista no assunto. Outra alternativa é procurar um centro médico em genética clínica mais próximo de você.
Acompanhamento multidisciplinar
O paciente com diagnóstico de MPS deve preferencialmente contar com o apoio de uma equipe multidisciplinar para que tenha um acompanhamento adequado às necessidades que a doença impõe.
Em razão das manifestações clínicas descritas em todos os tipos de MPS é importante o acompanhamento multidisciplinar, para prevenir e diagnosticar precocemente as complicações, que podem ser tratadas melhorando a qualidade de vida do portador de MPS e oferecendo apoio à família.
Geneticista clínico e neurologista
O acompanhamento periódico com intervalos de 4-6 meses, para controle do portador de MPS deve ser feito por geneticistas, pediatras ou clínicos gerais com especialização na área de erros inatos do metabolismo, que conheçam a evolução e as complicações possíveis da doença, para diagnosticar e encaminhar precocemente o paciente aos tratamentos necessários. Nos retornos os portadores de MPS devem receber avaliação clínica geral e neurológica, encaminhamentos para outras especialidades e pedidos de exames necessários ao seguimento.
Ortopedia e Radiologia
Pelo grande e progressivo comprometimento ósseo, o especialista deve ser consultado com intervalos regulares e no mínimo uma vez ao ano, se ainda não existirem alterações significativas. A realização de radiografias de coluna e ossos longos pelo menos uma vez ao ano é necessária para a avaliação de possíveis deformidades que, se diagnosticadas mais precocemente, podem ainda ser revertidas com posicionamento correto e fisioterapia específica.
Fisioterapia
Acompanhamento importante para a manutenção da qualidade de vida dos pacientes e para prevenir algumas complicações motoras e respiratórias (ver orientações). Nos pacientes que apresentam alterações de comportamento, principalmente os portadores de MPS III a eqüinoterapia (terapia na qual utiliza-se o convívio com os cavalos) tem se mostrado de grande ajuda.
Pneumologia/polissonografia
Devido à infiltração dos tecidos das vias aéreas superiores, a avaliação da capacidade pulmonar e polissonografia (que avalia a possibilidade de apnéia do sono estar interferindo na qualidade de vida do paciente) podem ser realizadas inicialmente uma vez ao ano e, caso indiquem algum tipo de intervenção, devem ser repetidas com uma freqüência específica em cada caso (ver orientações).
Otorrinolaringologia
A avaliação e procedimentos podem ser indicados e realizados por especialistas que tenham conhecimento da doença, pois em razão de suas peculiaridades, as cirurgias necessárias podem ser mais complicadas. Nesses casos, a anestesia geral é um procedimento de maior risco, principalmente pela dificuldade de entubação orotraqueal (devido à diminuição de espaço nas vias aéreas e pela chance de luxação atlanto-axial) que é necessária para manter as condições respiratórias durante qualquer procedimento cirúrgico.
Odontologia
Os cuidados devem ser mais intensos, pois com a hipertrofia das gengivas e prejuízo do esmalte dos dentes as chances de cáries e suas complicações são maiores (ver Orientações).
Fonoaudiologia
Avaliação da motricidade oral, da mastigação, deglutição e da fala, atenuando e prevenindo as complicações (vide orientações).
Psicologia
Apoio aos pais durante e após o diagnóstico para melhor compreensão das limitações do portador de MPS, evitando-se, porém, a super proteção e permitindo o desenvolvimento emocional. A psicoterapia aos portadores de MPS e/ou aos pais, com o objetivo de auxílio na superação das alterações que aconteceram na vida de ambos, em decorrência da doença, pode ser indicada quando necessária.
Cardiologia
Avaliação semestral, mas, se houver complicações, aumentar a freqüência de acordo com a necessidade. O ideal é que seja realizado o ecocardiograma a cada 6 meses. O médico é a pessoa adequada a orientar esse acompanhamento.
Oftalmologia
Além da existência de opacidade de córneas em alguns tipos, existem outras alterações correlacionadas, como a retinite pigmentar, disfunções retinianas, miopia e glaucoma. Assim o exame oftalmológico cuidadoso com lâmpada de fenda é aconselhável a cada 6 meses, a critério médico.
Neurocirurgia
O acompanhamento com este especialista pode ser indicado precocemente pelo médico que acompanha o paciente, quando notar alterações sugestivas da existência de hidrocefalia. A indicação de cirurgia deverá ser precisa e cuidadosa pelas dificuldades anestésicas já descritas, sendo também importante o acompanhamento por especialista que tenha experiência com MPS.
Recursos Adicionais
Os seguintes links são fornecidos como recursos adicionais de informações:
MPSIdisease.com foi criado para fornecer informações com relação a pacientes com MPS I, suas famílias, profissionais de saúde e de assistência médica. Aqui, você encontrará informações sobre a doença, programas de apoio e recursos on-line para administrar os desafios associados à MPS I. Conteúdo do site em inglês.
REFERÊNCIAS
1. Neufeld, E.F., e Muenzer, J. (2001) The mucopolysaccharidoses. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Childs, B., Kinzler, K.W., e Vogelstein, B. (eds.). 8th edition, Vol. III. McGraw-Hill, Medical Publishing Division





.jpg)














