A Esclerose Tuberosa é uma doença hereditária autossómica dominante com uma incidência de 1/10.000 nascimentos. Foram identificados dois loci principais: um em 9q34 (TSC1) e outro em 16p13 (TSC2). Ambos os genes são supressores tumorais. O gene TSC2 codifica para a tuberina. Mais de metade dos casos são esporádicos (mutações espontâneas). A penetrância atinge os 95% com uma expressividade muito variável, mesmo dentro da mesma família. Na idade adulta, 95% dos doentes apresentam lesões muito sugestivas: angiofibromas faciais, tumores de Koenen, placas fibrosas na fronte e couro cabeludo, angiomiolipomas renais, nódulos subependimários ou tuberomas corticais múltiplos e hamartomas da retina. Os sintomas podem ser muito discretos na infância. A epilepsia, habitualmente generalizada, é frequente (60% dos casos) e de difícil controlo. O atraso mental está presente em mais de 50% dos casos. O diagnóstico rigoroso é essencial de forma a detectar e tratar as lesões sintomáticas (neurológicas, renais, cardíacas e por vezes pulmonares), dado que são as principais causas de morte nestes doentes. As lesões faciais inestéticas ou problemáticas (angiofibromas faciais ou tumores de Koenen) podem ser removidos cirurgicamente ou por laser. O aconselhamento genético é difícil devido à grande variabilidade do fenótipo. *Autor: P. Wolkenstein, M.D. (Abril 2004)*.
VIGABATRINA NO TRATAMENTO DA EPILEPSIA DEDIFÍCIL CONTROLE EM PACIENTES COM SÍNDROMEDE WEST E ESCLEROSE TUBEROSA
Newra Tellechea Rotta
1, Alexandre Rodrigues da Silva
2, Lygia Ohlweiler
3, Rudimar Riesgo
3RESUMO - Objetivo é relatar a eficácia da vigabatrina no controle das convulsões, bem como as alterações eletrencefalográficas em crianças com esclerose tuberosa e síndrome de West. Método: Estudo retrospectivo,
com dados clínicos, de neuroimagem e de eletrencefalograma. Resultados: Sete pacientes foram acompanhados e o tempo médio de seguimento foi 10 anos. Dos pacientes, quatro eram do sexo feminino e todos eram de
cor branca. A média de idade de início das convulsões foi 3,4 meses. Todos usaram associações de vários anticonvulsivantes; no mínimo duas drogas por esquema terapêutico, e cada paciente utilizou pelo menos
dois esquemas diferentes. O uso de vigabatrina como monoterapia ou em associação iniciou em média aos 7 anos de idade ou 4 anos após início dos sintomas. Cinco dos sete pacientes que iniciaram vigabatrina ficaram
sem crise. Conclusão: Vigabatrina mostrou-se eficaz no controle das crises, levando a um melhor prognóstico.PALAVRAS-CHAVE: vigabatrina, esclerose tuberosa, síndrome de West, epilepsia.
Vigabatrin in the treatment of epilepsy in patients with West syndrome and tuberous sclerosis
A B S T R A C T - P u r p o s e : To r e p o r t t h e e f f i c a c y o f v i g a b a t r i n i n s e i z u r e s c o n t r o l , a s w e l l a s t h e
electroencephalographic abnormalities in children with tuberous sclerosis and West syndrome. Method:
Retrospective study, with clinical, neuroimaging, and electroencephalographic data. Results: Seven patients
were followed, and the median time of follow-up was 10 years. Four of them were females and all were
white. The mean age of seizures onset was 3.4 months. All patients used antiepileptic drugs associations, at
least 2 drugs each therapeutic scheme, each one of the patients have used at least two different schemes.
Vigabatrin as monotherapy or adjuvant was started in a mean age of seven years or 4 years after the onset of
symptons. Five from seven patients on vigabatrin became seizure free. Conclusion: Vigabatrin was efficient in
seizures control, leading to a better prognosis.
KEY WORDS: vigabatrin, tuberous sclerosis, West syndrome, epilepsy.
Unidade de Neurologia Pediátrica do Hospital de Clínicas de Porto Alegre, Porto Alegre RS, Brasil:
1
Livre Docente em Neurologia, Chefe
da Unidade de Neurologia Infantil;
2
Residente de Neurologia Infantil;
3
Neuropediatra, Doutor em Pediatria
Recebido 10 Fevereiro 2003, recebido na forma final 29 Maio 2003. Aceito 1 Julho 2003.
Dr. Alexandre Rodrigues da Silva - Rua Sofia Veloso 46/402 - 90050-140 Porto Alegre RS - Brasil. E-mail: alex164@ig.com.br
Espasmos infantis são o principal achado clínico
na síndrome de West, uma síndrome epilética relacionada com a idade que afeta 1 em cada 2000 a 4000
crianças
1
. O diagnóstico da síndrome de West é feito pela associação de episódios de espasmos em flexão, em extensão ou mistos, associados a EEG hipsarrítmico e, na maioria dos pacientes, retardo no
desenvolvimento neuropsicomotor. Sua etiologia pode variar desde causas infecciosas, tais como do grupo TORCH, passando por malformações cerebrais e
até causas idiopáticas. Esclerose tuberosa é um grupo de desordens autossômicas caracterizada por pela
presença de manchas hipocrômicas e tubérculos na
pele e por hamartomas e lesões neoplásicas que invariavelmente afetam o sistema nervoso central
2
. Pouco se sabe a respeito de suas bases fisiopatológicas
e o tratamento permanece problemático
3
. As principais alterações anatomopatológicas principais são
tubérculos corticais, nódulos gliais e subependimá-
rios e tumores de células gigantes
2,4
. Está frequentemente associada a síndrome de West, o que piora
tanto o prognóstico por suas consequências como
pela ocorrência da síndrome epiléptica
3,5
. Mesmo assim, possui características clínicas e eletrográficas distintas da síndrome de West que ocorre por outras
etiologias
4
.
O objetivo deste estudo é relatar a eficácia da
vigabatrina no controle das crises convulsivas e nas
alterações eletrencefalográficas em pacientes com
esclerose tuberosa e síndrome de West refratáriosArq Neuropsiquiatr 2003;61(4) 989
ao tratamento com outros anticonvulsivantes, seja
em mono ou politerapia.
MÉTODO
Foram selecionados 7 casos de esclerose tuberosa e
síndrome de West de um universo de 84 pacientes com
síndrome de West do ambulatório de Neurologia Infantil
do Hospital de Clínicas de Porto Alegre. Foram incluídos
pacientes de 0 a 12 anos com diagnóstico de esclerose
tuberosa de acordo com os critérios diagnósticos estabelecidos pelo Comitê de Critérios da Associação Americana
de Esclerose Tuberosa e que apresentavam também quadro compatível com síndrome de West
2,4
.
Até o controle das crises, os pacientes foram acompanhados de três em três meses, e após, semestralmente,
tendo sido submetidos de forma periódica a exames clínicos, laboratoriais, eletrencefalográficos e de neuroimagem.
Avaliou-se a evolução clínica, a partir dos esquemas de
ant i convul s i vantes ut i l i zados , bem como o uso de
vigabatrina, tanto em monoterapia, como em terapia
adjuvante.
RESULTADOS
Sete pacientes com esclerose tuberosa e síndrome
de West tiveram seguimento ambulatorial regular e
usaram vigabatrina. A Tabela 1 mostra os dados demográficos desta amostra. A Tabela 2 mostra o resultado clínico do uso de vigabatrina. Desse grupo,
quatro pacientes eram do sexo feminino e todos
eram de cor branca. Os sintomas iniciais foram de
espasmos em flexão dos membros em 4 pacientes e
em extensão nos demais. A média de idade de início
dos sintomas foi 3,4 meses (variando de 2 a 8 meses), e o achado eletrencefalográfico inicial foi hipsarritmia. Em todos os casos os exames de tomografia computadorizada e ressonância nuclear magnética de encéfalo foram compatíveis com esclerose
tuberosa. O tempo médio de acompanhamento
ambulatorial foi 10 anos (variando de 27 meses a
17 anos). Na avaliação neurológica, 5 pacientes apresentavam retardo do desenvolvimento neuropsicomotor, sendo que um deles apresentou evolução
compatível com síndrome de Lennox-Gastaut no
decorrer do acompanhamento (paciente 5), um desenvolveu conduta dentro do espectro do autismo
(paciente 1) e outro apresentou sintomas psicóticos
(paciente 7).
A história familiar era positiva para epilepsia em
2 casos, mas em nenhum havia diagnóstico de esclerose tuberosa. Todos apresentavam variados graus
de comprometimento cutâneo, desde tubérculos na
face a manchas hipocrômicas no tronco ou a associação de ambas. Um paciente teve um astrocitoma
subependimário de células gigantes. Todos usaram
esquema com vários anticonvulsivantes; no mínimo
2 drogas por esquema terapêutico, sendo que cada
paciente utilizou pelo menos 2 esquemas diferentes, que incluíam ácido valpróico, carbamazepina,
oxacarbamazepina, fenobarbital, primidona, nitrazepan e corticóides. O uso de vigabatrina como
monoterapia ou adjuvante a outros anticonvulsivantes iniciou em média aos 7 anos (variação de 2 -
17 anos) ou 42 meses após início dos sintomas (varia-
ção de 20 – 82 meses). Os pacientes que iniciaram
vigabatrina tiveram pronta melhora das crises, tanto em quantidade como em intensidade e duração.
Cinco dos 7 pacientes que iniciaram vigabatrina ficaram sem crise. Um caso (paciente 4) no qual não
houve melhora completa das crises tinha tumor cerebral e foi o de início mais tardio do uso da vigabatrina
- 82 meses -, tendo sido tentados três esquemas de
anticonvulsivantes anteriormente. Após o início da
vigabatrina, as crises, que eram de frequência diária
(3-4/dia), começaram se tornar semanais, com menor duração cada uma.
Tabela 1. Dados demográficos.
Paciente Sexo Início História familiar Tempo de
dos sintomas seguimento
(meses) (meses)
1 F 8 5 primos com epilepsia 204
2 F 8 Irmão com hamartoma de hipófise 27
3 F 4 - 164
4 M 3 - 182
5 F 3 2 tios paternos com epilepsia 63
6 M 3 - 47
7 M 2 Primo com síndrome de Down 130990 Arq Neuropsiquiatr 2003;61(4)
Três pacientes evoluíram com EEG mostrando alterações focais temporais, um frontal bilateral, e dois
multifocais, além de um caso que evoluiu para EEG
compatível com síndrome de Lennox-Gastaut.
DISCUSSÃO
Vários estudos têm demonstrado a eficácia da vigabatrina como monoterapia ou como fármaco adjuvante no tratamento da esclerose tuberosa associada
a síndrome de West sendo bem tolerada e com poucos efeitos adversos
6-11
. No entanto, um estudo de
meta análise
2
não encontrou evidência de que a vigabatrina tivesse algum tipo de vantagem sobre as outras opções terapêuticas. Em casos refratários ao uso
de vigabatrina e ACTH, opções como piridoxina, lamotrigina, topiramato, zonisamida, dieta cetogênica,
terapia com imunoglobulina, felbamato e hormônio
liberador de tireotrofina têm sido relatadas
1
.
No estudo de Husain
3
, uma série de 17 casos de
esclerose tuberosa e síndrome de West na qual foi
usada a vigabatrina, observou-se uma melhora estatisticamente significativa do padrão do EEG, com
melhor controle das crises. Os autores concluíram
que o EEG é o melhor indicador para o controle das
crises. Outro fator de impacto prognóstico é a localização e o tamanho dos tubérculos intracerebrais
4,8,12
,
sendo que os episódios convulsivos mais precoces
ocorrem quando os tubérculos estão localizados nas
regiões posteriores. Não conseguimos estabelecer
essa relação no nosso estudo. O estudo de Gonzalez
13
correlaciona os casos de síndrome de West associados à esclerose tuberosa com uma maior prevalência
de autismo, o que foi encontrado em um dos nossos pacientes. Concordamos que nesses casos de
Tabela 2. Tratamento anticonvulsivante.
Pacientes Esquema de Padrão EEG Pré- Início da Uso Estado pós- Tempo de
pré-vigabatrina vigabatrina vigabatrina prévio de vigabatrina Acompanhamento
corticóide pós-vigabatrina
1 VPA+CBZ+FNB Parox Temporal D 17 anos + sem crises 2 anos
2 CBZ+VPA Parox Frontal bilateral 34 meses - sem crises 1 ano
3 VPA+CBZ+primidona Parox Multifocal - + -
4 CBZ+VPA Parox Temporal E 9 anos + - crises 8 anos
5 FNB+VPA+nitrazepam Lennox-Gastaut 23 meses - sem crises 7 anos
6 FNB+VPA Parox Temporal D 47 meses - sem crises 7 anos
7 FNB + CBZ Parox Multifocal 7 anos - sem crises 4 anos
F, frontal; T, temporal; D, direito; E, esquerdo.
esclerose tuberosa associados a síndrome de West
deve-se sempre suspeitar dessa disfunção neurocomportamental, de maneira a proporcionar estimulação
especial. Em conclusão, a vigabatrina mostrou-se eficaz no controle das crises convulsivas, nos casos de
síndrome de West associada à esclerose tuberosa,
do que resultou melhor prognóstico. Deve-se buscar sempre uma avaliação pormenorizada através do
exame neurológico, de neuroimagem e eletrencefalográfico para melhor estabelecer o tratamento farmacológico e o prognóstico desses casos.
REFERÊNCIAS
1. Mikati MA, Lepejian GA, Holmes GL. Medical treatment of patients
with infantile spasms. Clin Neuropharmacol 2002;25:61-70.
2. Berg BO . Neurocutanous syndromes: phacomatosis and alliend
conditions. In Swaiman KF (ed.) Pediatric neurology. principles and
practice. St. Louis Mosby, 1989;795-797.
3. Reis-Filho JS, Montemor Netto MR, Loyola Netto JG, de Araujo JC,
An t o n i u k S , T o r r e s L F . [ T u b e r o u s s c l e r o s i s : c a s e r e p o r t wi t h
histopathological and ultrastructural study]. Arq Neuropsiquiatr
1998;56:671-676.
4. Gomez MR. Tuberous sclerosis. New York Raven Press, 1979.
5. Hancock E, Osborne JP, Milner P. Treatment of infantile spasms
(Cochrane Review). Cochrane Database Syst Rev 2002;(2):CD001770.
6. Husain AM, Foley CM, Legido A, Chandler DA, Miles DK, Grover
WD.West syndrome in tuberous sclerosis complex. Pediatr Neurol
2000;23:233-235.
7. Curatolo P, Seri S, Verdecchia M, Bombardieri R. Infantile spasms in
tuberous sclerosis complex. Brain Dev 2001;23:502-507.
8. Lopez-Valdes E, Hernandez-Lain A, Simon R, Porta J, Mateos F.
[Treatment of refractory infantile epilepsy with vigabatrin in a series
of 55 patients]. Rev Neurol (Barc) 1996;24:1255-1257.
9. Rufo M, Santiago C, Castro E, Ocana O. [Monotherapy with vigabatrin in
the treatment of West’s syndrome]. Rev Neurol (Barc) 1997;25:1365-1368.
10. Hamano S, Tanaka M, Imai M, Nara T, Maekawa K. [Topography, and
number of cortical tubers in tuberous sclerosis: comparison between patients
with and without West syndrome] No To Hattatsu 1999;31:402-407.
11. Chiron C. [Role of physiopathological hypotheses in therapeutic choice
in epilepsy in children] : Rev Neurol (Paris) 1997;153 Suppl 1:S34-38.
12. Ohtsuka Y, Ohmori I, Oka E.Long-term follow-up of childhood epilepsy
associated with tuberous sclerosis. Epilepsia 1998;39:1158-1163.
13. Calderón González R, Treviño Welsh J; Calderón Sepúlveda A. Autismo
en la esclerosis tuberosa. Gac Méd Méx 1994;130:374-379.
VIGABATRINA NO TRATAMENTO DA EPILEPSIA DEDIFÍCIL CONTROLE EM PACIENTES COM SÍNDROMEDE WEST E ESCLEROSE TUBEROSA
Newra Tellechea Rotta
1, Alexandre Rodrigues da Silva
2, Lygia Ohlweiler
3, Rudimar Riesgo
3RESUMO - Objetivo é relatar a eficácia da vigabatrina no controle das convulsões, bem como as alterações eletrencefalográficas em crianças com esclerose tuberosa e síndrome de West. Método: Estudo retrospectivo,
com dados clínicos, de neuroimagem e de eletrencefalograma. Resultados: Sete pacientes foram acompanhados e o tempo médio de seguimento foi 10 anos. Dos pacientes, quatro eram do sexo feminino e todos eram de
cor branca. A média de idade de início das convulsões foi 3,4 meses. Todos usaram associações de vários anticonvulsivantes; no mínimo duas drogas por esquema terapêutico, e cada paciente utilizou pelo menos
dois esquemas diferentes. O uso de vigabatrina como monoterapia ou em associação iniciou em média aos 7 anos de idade ou 4 anos após início dos sintomas. Cinco dos sete pacientes que iniciaram vigabatrina ficaram
sem crise. Conclusão: Vigabatrina mostrou-se eficaz no controle das crises, levando a um melhor prognóstico.PALAVRAS-CHAVE: vigabatrina, esclerose tuberosa, síndrome de West, epilepsia.
Vigabatrin in the treatment of epilepsy in patients with West syndrome and tuberous sclerosis
A B S T R A C T - P u r p o s e : To r e p o r t t h e e f f i c a c y o f v i g a b a t r i n i n s e i z u r e s c o n t r o l , a s w e l l a s t h e
electroencephalographic abnormalities in children with tuberous sclerosis and West syndrome. Method:
Retrospective study, with clinical, neuroimaging, and electroencephalographic data. Results: Seven patients
were followed, and the median time of follow-up was 10 years. Four of them were females and all were
white. The mean age of seizures onset was 3.4 months. All patients used antiepileptic drugs associations, at
least 2 drugs each therapeutic scheme, each one of the patients have used at least two different schemes.
Vigabatrin as monotherapy or adjuvant was started in a mean age of seven years or 4 years after the onset of
symptons. Five from seven patients on vigabatrin became seizure free. Conclusion: Vigabatrin was efficient in
seizures control, leading to a better prognosis.
KEY WORDS: vigabatrin, tuberous sclerosis, West syndrome, epilepsy.
Unidade de Neurologia Pediátrica do Hospital de Clínicas de Porto Alegre, Porto Alegre RS, Brasil:
1
Livre Docente em Neurologia, Chefe
da Unidade de Neurologia Infantil;
2
Residente de Neurologia Infantil;
3
Neuropediatra, Doutor em Pediatria
Recebido 10 Fevereiro 2003, recebido na forma final 29 Maio 2003. Aceito 1 Julho 2003.
Dr. Alexandre Rodrigues da Silva - Rua Sofia Veloso 46/402 - 90050-140 Porto Alegre RS - Brasil. E-mail: alex164@ig.com.br
Espasmos infantis são o principal achado clínico
na síndrome de West, uma síndrome epilética relacionada com a idade que afeta 1 em cada 2000 a 4000
crianças
1
. O diagnóstico da síndrome de West é feito pela associação de episódios de espasmos em flexão, em extensão ou mistos, associados a EEG hipsarrítmico e, na maioria dos pacientes, retardo no
desenvolvimento neuropsicomotor. Sua etiologia pode variar desde causas infecciosas, tais como do grupo TORCH, passando por malformações cerebrais e
até causas idiopáticas. Esclerose tuberosa é um grupo de desordens autossômicas caracterizada por pela
presença de manchas hipocrômicas e tubérculos na
pele e por hamartomas e lesões neoplásicas que invariavelmente afetam o sistema nervoso central
2
. Pouco se sabe a respeito de suas bases fisiopatológicas
e o tratamento permanece problemático
3
. As principais alterações anatomopatológicas principais são
tubérculos corticais, nódulos gliais e subependimá-
rios e tumores de células gigantes
2,4
. Está frequentemente associada a síndrome de West, o que piora
tanto o prognóstico por suas consequências como
pela ocorrência da síndrome epiléptica
3,5
. Mesmo assim, possui características clínicas e eletrográficas distintas da síndrome de West que ocorre por outras
etiologias
4
.
O objetivo deste estudo é relatar a eficácia da
vigabatrina no controle das crises convulsivas e nas
alterações eletrencefalográficas em pacientes com
esclerose tuberosa e síndrome de West refratáriosArq Neuropsiquiatr 2003;61(4) 989
ao tratamento com outros anticonvulsivantes, seja
em mono ou politerapia.
MÉTODO
Foram selecionados 7 casos de esclerose tuberosa e
síndrome de West de um universo de 84 pacientes com
síndrome de West do ambulatório de Neurologia Infantil
do Hospital de Clínicas de Porto Alegre. Foram incluídos
pacientes de 0 a 12 anos com diagnóstico de esclerose
tuberosa de acordo com os critérios diagnósticos estabelecidos pelo Comitê de Critérios da Associação Americana
de Esclerose Tuberosa e que apresentavam também quadro compatível com síndrome de West
2,4
.
Até o controle das crises, os pacientes foram acompanhados de três em três meses, e após, semestralmente,
tendo sido submetidos de forma periódica a exames clínicos, laboratoriais, eletrencefalográficos e de neuroimagem.
Avaliou-se a evolução clínica, a partir dos esquemas de
ant i convul s i vantes ut i l i zados , bem como o uso de
vigabatrina, tanto em monoterapia, como em terapia
adjuvante.
RESULTADOS
Sete pacientes com esclerose tuberosa e síndrome
de West tiveram seguimento ambulatorial regular e
usaram vigabatrina. A Tabela 1 mostra os dados demográficos desta amostra. A Tabela 2 mostra o resultado clínico do uso de vigabatrina. Desse grupo,
quatro pacientes eram do sexo feminino e todos
eram de cor branca. Os sintomas iniciais foram de
espasmos em flexão dos membros em 4 pacientes e
em extensão nos demais. A média de idade de início
dos sintomas foi 3,4 meses (variando de 2 a 8 meses), e o achado eletrencefalográfico inicial foi hipsarritmia. Em todos os casos os exames de tomografia computadorizada e ressonância nuclear magnética de encéfalo foram compatíveis com esclerose
tuberosa. O tempo médio de acompanhamento
ambulatorial foi 10 anos (variando de 27 meses a
17 anos). Na avaliação neurológica, 5 pacientes apresentavam retardo do desenvolvimento neuropsicomotor, sendo que um deles apresentou evolução
compatível com síndrome de Lennox-Gastaut no
decorrer do acompanhamento (paciente 5), um desenvolveu conduta dentro do espectro do autismo
(paciente 1) e outro apresentou sintomas psicóticos
(paciente 7).
A história familiar era positiva para epilepsia em
2 casos, mas em nenhum havia diagnóstico de esclerose tuberosa. Todos apresentavam variados graus
de comprometimento cutâneo, desde tubérculos na
face a manchas hipocrômicas no tronco ou a associação de ambas. Um paciente teve um astrocitoma
subependimário de células gigantes. Todos usaram
esquema com vários anticonvulsivantes; no mínimo
2 drogas por esquema terapêutico, sendo que cada
paciente utilizou pelo menos 2 esquemas diferentes, que incluíam ácido valpróico, carbamazepina,
oxacarbamazepina, fenobarbital, primidona, nitrazepan e corticóides. O uso de vigabatrina como
monoterapia ou adjuvante a outros anticonvulsivantes iniciou em média aos 7 anos (variação de 2 -
17 anos) ou 42 meses após início dos sintomas (varia-
ção de 20 – 82 meses). Os pacientes que iniciaram
vigabatrina tiveram pronta melhora das crises, tanto em quantidade como em intensidade e duração.
Cinco dos 7 pacientes que iniciaram vigabatrina ficaram sem crise. Um caso (paciente 4) no qual não
houve melhora completa das crises tinha tumor cerebral e foi o de início mais tardio do uso da vigabatrina
- 82 meses -, tendo sido tentados três esquemas de
anticonvulsivantes anteriormente. Após o início da
vigabatrina, as crises, que eram de frequência diária
(3-4/dia), começaram se tornar semanais, com menor duração cada uma.
Tabela 1. Dados demográficos.
Paciente Sexo Início História familiar Tempo de
dos sintomas seguimento
(meses) (meses)
1 F 8 5 primos com epilepsia 204
2 F 8 Irmão com hamartoma de hipófise 27
3 F 4 - 164
4 M 3 - 182
5 F 3 2 tios paternos com epilepsia 63
6 M 3 - 47
7 M 2 Primo com síndrome de Down 130990 Arq Neuropsiquiatr 2003;61(4)
Três pacientes evoluíram com EEG mostrando alterações focais temporais, um frontal bilateral, e dois
multifocais, além de um caso que evoluiu para EEG
compatível com síndrome de Lennox-Gastaut.
DISCUSSÃO
Vários estudos têm demonstrado a eficácia da vigabatrina como monoterapia ou como fármaco adjuvante no tratamento da esclerose tuberosa associada
a síndrome de West sendo bem tolerada e com poucos efeitos adversos
6-11
. No entanto, um estudo de
meta análise
2
não encontrou evidência de que a vigabatrina tivesse algum tipo de vantagem sobre as outras opções terapêuticas. Em casos refratários ao uso
de vigabatrina e ACTH, opções como piridoxina, lamotrigina, topiramato, zonisamida, dieta cetogênica,
terapia com imunoglobulina, felbamato e hormônio
liberador de tireotrofina têm sido relatadas
1
.
No estudo de Husain
3
, uma série de 17 casos de
esclerose tuberosa e síndrome de West na qual foi
usada a vigabatrina, observou-se uma melhora estatisticamente significativa do padrão do EEG, com
melhor controle das crises. Os autores concluíram
que o EEG é o melhor indicador para o controle das
crises. Outro fator de impacto prognóstico é a localização e o tamanho dos tubérculos intracerebrais
4,8,12
,
sendo que os episódios convulsivos mais precoces
ocorrem quando os tubérculos estão localizados nas
regiões posteriores. Não conseguimos estabelecer
essa relação no nosso estudo. O estudo de Gonzalez
13
correlaciona os casos de síndrome de West associados à esclerose tuberosa com uma maior prevalência
de autismo, o que foi encontrado em um dos nossos pacientes. Concordamos que nesses casos de
Tabela 2. Tratamento anticonvulsivante.
Pacientes Esquema de Padrão EEG Pré- Início da Uso Estado pós- Tempo de
pré-vigabatrina vigabatrina vigabatrina prévio de vigabatrina Acompanhamento
corticóide pós-vigabatrina
1 VPA+CBZ+FNB Parox Temporal D 17 anos + sem crises 2 anos
2 CBZ+VPA Parox Frontal bilateral 34 meses - sem crises 1 ano
3 VPA+CBZ+primidona Parox Multifocal - + -
4 CBZ+VPA Parox Temporal E 9 anos + - crises 8 anos
5 FNB+VPA+nitrazepam Lennox-Gastaut 23 meses - sem crises 7 anos
6 FNB+VPA Parox Temporal D 47 meses - sem crises 7 anos
7 FNB + CBZ Parox Multifocal 7 anos - sem crises 4 anos
F, frontal; T, temporal; D, direito; E, esquerdo.
esclerose tuberosa associados a síndrome de West
deve-se sempre suspeitar dessa disfunção neurocomportamental, de maneira a proporcionar estimulação
especial. Em conclusão, a vigabatrina mostrou-se eficaz no controle das crises convulsivas, nos casos de
síndrome de West associada à esclerose tuberosa,
do que resultou melhor prognóstico. Deve-se buscar sempre uma avaliação pormenorizada através do
exame neurológico, de neuroimagem e eletrencefalográfico para melhor estabelecer o tratamento farmacológico e o prognóstico desses casos.
REFERÊNCIAS
1. Mikati MA, Lepejian GA, Holmes GL. Medical treatment of patients
with infantile spasms. Clin Neuropharmacol 2002;25:61-70.
2. Berg BO . Neurocutanous syndromes: phacomatosis and alliend
conditions. In Swaiman KF (ed.) Pediatric neurology. principles and
practice. St. Louis Mosby, 1989;795-797.
3. Reis-Filho JS, Montemor Netto MR, Loyola Netto JG, de Araujo JC,
An t o n i u k S , T o r r e s L F . [ T u b e r o u s s c l e r o s i s : c a s e r e p o r t wi t h
histopathological and ultrastructural study]. Arq Neuropsiquiatr
1998;56:671-676.
4. Gomez MR. Tuberous sclerosis. New York Raven Press, 1979.
5. Hancock E, Osborne JP, Milner P. Treatment of infantile spasms
(Cochrane Review). Cochrane Database Syst Rev 2002;(2):CD001770.
6. Husain AM, Foley CM, Legido A, Chandler DA, Miles DK, Grover
WD.West syndrome in tuberous sclerosis complex. Pediatr Neurol
2000;23:233-235.
7. Curatolo P, Seri S, Verdecchia M, Bombardieri R. Infantile spasms in
tuberous sclerosis complex. Brain Dev 2001;23:502-507.
8. Lopez-Valdes E, Hernandez-Lain A, Simon R, Porta J, Mateos F.
[Treatment of refractory infantile epilepsy with vigabatrin in a series
of 55 patients]. Rev Neurol (Barc) 1996;24:1255-1257.
9. Rufo M, Santiago C, Castro E, Ocana O. [Monotherapy with vigabatrin in
the treatment of West’s syndrome]. Rev Neurol (Barc) 1997;25:1365-1368.
10. Hamano S, Tanaka M, Imai M, Nara T, Maekawa K. [Topography, and
number of cortical tubers in tuberous sclerosis: comparison between patients
with and without West syndrome] No To Hattatsu 1999;31:402-407.
11. Chiron C. [Role of physiopathological hypotheses in therapeutic choice
in epilepsy in children] : Rev Neurol (Paris) 1997;153 Suppl 1:S34-38.
12. Ohtsuka Y, Ohmori I, Oka E.Long-term follow-up of childhood epilepsy
associated with tuberous sclerosis. Epilepsia 1998;39:1158-1163.
13. Calderón González R, Treviño Welsh J; Calderón Sepúlveda A. Autismo
en la esclerosis tuberosa. Gac Méd Méx 1994;130:374-379.
| ESCLEROSE TUBEROSA | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Doença rara e pouco conhecida de difícil diagnóstico. Em muitos casos de Esclerose Tuberosa os portadores são tardiamente diagnosticados e a falta de informação dificulta o tratamento de seus sintomas. A Esclerose Tuberosa, também conhecida como Síndrome de Bourneville-Pringle ou Epilóia, é uma desordem genética e, portanto, uma doença não contagiosa, causada por anomalias nos genes TSC1 ou TSC2, dos cromossomos 9 e 16, respectivamente. Inicialmente a Esclerose Tuberosa foi descrita no sistema nervoso central por Bourneville, em 1880. É uma doença degenerativa, causadora de tumores benignos, que pode afetar diversos órgãos, especialmente cérebro, coração, olhos, rins, pele e pulmões. As manifestações clínicas da doença podem variar dependendo do grau de acometimento dos órgãos afetados. Podendo surgir lesões na pele, nos ossos, dentes, rins, pulmões, olhos, coração e sistema nervoso central. As lesões dermatológicas se apresentam sob a forma de nódulos de cor vermelha ou cereja geralmente na região facial. As lesões retinianas afetam as camadas superficiais da retina e as lesões cerebrais podem ser tumores e calcificações na região dos ventrículos cerebrais. É uma doença rara, de tendência evolutiva que pode afetar ambos os sexos de todas as raças e grupos étnicos.Ao primeiro sinal de depressão, os pais devem acolher a criança e encaminhá-la a um profissional o mais rápido possível. Na maioria das vezes, o apoio da família e a psicoterapia são suficientes. Somente a partir dos 6 anos de idade, é necessário, em alguns casos, intervir com medicamentos. A depressão infantil desencadeia várias outras doenças tais como: anorexia, bulimia, etc.  A Esclerose Tuberosa é caracterizada por Deficiência Mental, epilepsia e erupção facial tipo adenoma sebáceo. Devido a tais características a doença também foi chamada de epilóia. epil = epilepsia oi = oligofrenia ==> Epiloia a = adenoma Os sintomas da Esclerose Tuberosa são o aparecimento de neoplasias benignas em um ou mais órgãos, sendo o sistema nervoso central e retina, pele, coração e rins os mais comuns de serem afetados. Tumores cerebrais no Sistema Nervoso Central também podem ocorrer em casos confirmados da doença, aparecendo até aos 20 anos de idade do paciente. Esclerose Tuberosa aparece no sistema nervoso central através de tumores benignos e nódulos. As variações de sintomas entre pacientes são muitas, mas, os mais comuns são as crises epiléticas, incluindo também o Retardo Mental ou a hidrocefalia secundária. A Deficiência Mental pode estar ausente em cerca de 30% dos casos, mas as convulsões, de difícil controle, aparecem em cerca de 80%, começando antes dos 5 anos de idade. Os exames que se pode fazer para constatação e acompanhamentos da Esclerose Tuberosos são os exames genéticos de Cariótipo, a Tomografia Craniana Computadorizada, Ressonância Magnética Nuclear e Craniografia. 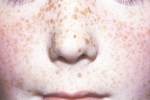 Ainda não há cura para essa síndrome, apenas tratamento para seus sintomas, mas há muito a ser feito para melhorar a qualidade de vida do portador. O tratamento é sintomático, isto é, procura-se sanar os sintomas que se manifestam, e normalmente é acompanhado da aplicação de anticonvulsivantes no intuito de se controlar as crises convulsivas presentes na maioria dos casos. Além das medicações convencionais, diversas formas de terapias já mostraram resultados positivos, como o shiatsu, o reiki e as freqüências de brilho que trazem benefícios que variam de quadro para quadro, mas podem ser sensíveis em alguns casos. Terapias como a Homeopatia, a Fisioterapia e a Terapia Ocupacional têm sido um ganho enorme junto aos pacientes portadores da doença. 
| ||||||||||||||




Nenhum comentário:
Postar um comentário
DEIXE SEU COMENTARIO E REDIRECIONE PARA O EMAIL PRESIDENCIAZN@YAHOO.COM.BR